
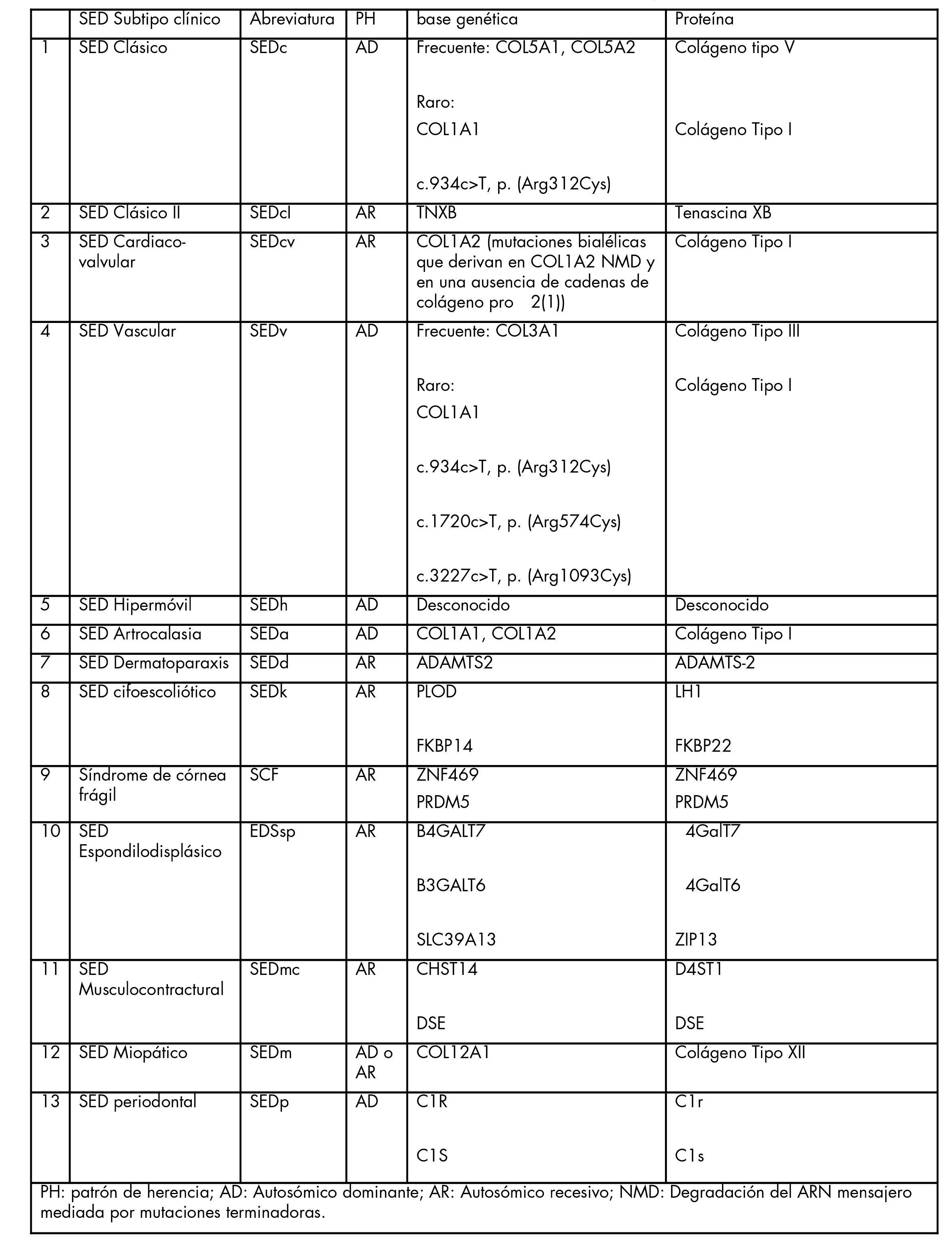
Clasificación internacional 2017 de los síndromes de Ehlers-Danlos
Los síndromes de Ehlers-Danlos (SED) son un grupo clínica y genéticamente heterogéneo de trastornos hereditarios del tejido conectivo (THTC) caracterizados por hipermovilidad de las articulaciones, hiperextensibilidad de la piel y fragilidad de los tejidos.
En las últimas dos décadas, la Nosología de Villefranche, que delineó seis subtipos, ha sido ampliamente utilizada como el estándar para el diagnóstico clínico de SED. Para la mayoría de estos subtipos, las mutaciones se han identificado en los genes que codifican el colágeno, o en los genes que codifican enzimas modificadoras del colágeno. Desde su publicación en 1998, se ha descrito todo un espectro de nuevos subtipos de SED, y se han identificado mutaciones en toda una serie de nuevos genes.
El Consorcio Internacional SED propone una clasificación SED revisada, que reconoce 13 subtipos. Para cada uno de los subtipos, proponemos un conjunto de criterios clínicos que son indicativos para el diagnóstico. Sin embargo, en vista de la gran heterogeneidad genética y la variabilidad fenotípica de los subtipos de SED, y la superposición clínica entre los subtipos de SED, también con otros TTC, el diagnóstico definitivo de todos los subtipos de SED, excepto para el tipo hipermóvil, se basa en la confirmación molecular con identificación de la/s variante/s genética/s causante/s. También revisamos los criterios clínicos para SED hipermóvil con el fin de permitir una mejor distinción de otros trastornos de hipermovilidad articular. Para satisfacer las necesidades de investigación, también se propone un esquema patogenético, que agrupa subtipos de SED en los que las proteínas causantes funcionan dentro de la misma vía.
Esperamos que la Clasificación Internacional SED revisada sirva como una nueva norma para el diagnóstico de SED y proporcione un marco para futuros propósitos de investigación. © 2017 Wiley Periodicals, Inc.
INTRODUCCIÓN
Los síndromes de Ehlers-Danlos (SED) son un grupo heterogéneo de trastornos hereditarios del tejido conectivo (THTC) caracterizados por hipermovilidad articular, hiperextensibilidad de la piel y fragilidad de los tejidos. La heterogeneidad clínica y genética de esta afección ha sido reconocida desde hace tiempo. La «Nosología de Berlín» de 1988 reconoció 11 subtipos, definidos por números romanos, basados en hallazgos clínicos y en el modo de herencia [Beighton et al., 1988]. La interpretación subjetiva de varios signos clínicos semicuantitativos, tales como la hipermovilidad articular, la hiperextensibilidad de la piel, la fragilidad de los tejidos y los moretones, sin embargo, dio lugar a la incertidumbre clínica, confusión diagnóstica con respecto al tipo de SED y la inclusión de condiciones fenotípicamente similares bajo el amplio diagnóstico de SED. Con la dilucidación de las bases bioquímicas y moleculares de muchos de estos tipos de SED, se publicó en 1998 una clasificación revisada, la «Nosología Villefranche» [Beighton et al., 1998]. Esta clasificación delineó seis subtipos, para los que se definieron criterios clínicos mayores y menores, los cuales incluyeron la base bioquímica y molecular, cuando eran conocidas. Los números romanos fueron sustituidos por un nombre descriptivo, que capturó las manifestaciones características de cada tipo. Una suposición subyacente era que la mayoría, si no todos, de estos tipos de SED eran consecuencia de alteraciones en los genes del colágeno fibrilar o en los genes que codificaban los modificadores del colágeno.
Con la elucidación de las bases bioquímicas y moleculares de muchos de estos tipos de SED, se publicó en 1998 una clasificación revisada, la «Nosología de Villefranche». Esta clasificación delineó seis subtipos, para los cuales se definieron criterios clínicos mayores y menores, que incluyeron la base bioquímica y molecular, cuando eran conocidas.
En las dos últimas décadas, la Nosología de Villefranche ha servido a su propósito y ha sido ampliamente utilizada como el estándar para el diagnóstico clínico de SED, y para la investigación clínica en varios aspectos de estas condiciones. Sin embargo, desde su publicación, se ha descrito todo un espectro de nuevos subtipos de SED, y con el llegada de las prestaciones de secuenciación de próxima generación (NSG por sus siglas en inglés), las mutaciones se han identificado en una serie de nuevos genes, que no siempre están, a primera vista , implicados en la biosíntesis y/o estructura del colágeno. Como tal, la clasificación de Villefranche ha quedado obsoleta. Además, en la falta persistente de un defecto genético, existe una seria necesidad de una mejor definición clínica del tipo hipermóvil de SED y su diferenciación de otros trastornos de hipermovilidad. Por lo tanto, realizamos una revisión exhaustiva de la literatura relacionada con el SED y, sobre la base de nuestros hallazgos, revisamos la Clasificación SED.
CLASIFICACIÓN INTERNACIONAL 2017 PARA LOS SÍNDROMES EHLERS-DANLOS
La nueva clasificación reconoce 13 subtipos, como se indica en la Tabla I. Después de cuidadosas deliberaciones sobre si mantener una clasificación clínicamente orientada versus una clasificación genética, proponemos mantener una clasificación clínica, en la cual se mantienen los nombres descriptivos previamente establecidos, ya que son generalmente aceptados y ampliamente utilizados en la comunidad médica, científica y del paciente. Para los nuevos fenotipos SED, se propone un nuevo descriptor que captura las manifestaciones características del fenotipo.
Se incluyeron todos los fenotipos que presentan las características clínicas básicas del SED, es decir, la hipermovilidad de las articulaciones, la hiperextensibilidad de la piel y la fragilidad de los tejidos. En particular, tales características deberían distinguir el tipo hipermóvil (SED hipermóvil, SEDh) de otros trastornos de hipermovilidad articular (véase también «Un marco para la clasificación de hipermovilidad articular y afecciones relacionadas» de Castori et al., en este número). Algunos de los fenotipos se superponen clínicamente con otros trastornos del tejido conectivo (TTC), como el «SED miopático», que es causado por mutaciones heterocigóticas o bialélicas en COL12A1 (SEDm) y que se superpone clínicamente con la miopatía de Bethlem y «SED espondilodisplástico» causado por mutaciones B3GALT6 bialélicas -B3GALT6), que se superpone clínicamente con la displasia espondilo-epimetafisaria con laxitud articular tipo I (SEMD-JL1). Dado que se sospecha clínicamente que varios pacientes con estas afecciones tienen una forma de SED, creemos que la inclusión en la clasificación SED está justificada. Este es también el caso del síndrome de córnea frágil. Actualmente no conservamos la heterotopía nodular periventricular relacionada con filamina A (HNPV) con características de SED dentro de la clasificación, ya que la mayoría de los pacientes presentan principalmente un fenotipo neurológico. Una minoría de pacientes tiene características diferentes de un TTC, que puede incluir aneurismas potencialmente letales, sin embargo, hay datos publicados insuficientes para diferenciar y pronosticar de forma fiable HNPV de HNPV-SED.
Se recomienda que en los años venideros se examine la inclusión o la exclusión de estas condiciones en la clasificación de SED, cuando se disponga de más información.
En línea con la Nosología de Villefranche 1997, se proponen un conjunto de criterios clínicos mayores y menores para cada subtipo de SED. Un criterio mayor tiene alta especificidad diagnóstica porque está presente en la gran mayoría de los individuos afectados y/o es característico para el trastorno y permite la diferenciación de otros subtipos de SED y/u otros TTC. Un criterio menor es un signo de menor especificidad diagnóstica, pero su presencia apoya el diagnóstico. Para cada uno de los subtipos, se definen los criterios clínicos mínimos menores ± menores que son sugerentes para el diagnóstico de un subtipo específico. Sin embargo, en vista de la gran heterogeneidad genética y la variabilidad fenotípica de los subtipos de SED, y la superposición clínica entre muchos de estos subtipos, pero también con otros TTC, el diagnóstico definitivo se basa en todos los subtipos, excepto SEDh, en la confirmación molecular con la identificación de variante/s causante/s en el gen respectivo. Un diagnóstico molecular es extremadamente importante para el asesoramiento, ya que permite confirmar el diagnóstico preciso y proporciona información sobre el patrón de herencia, el riesgo de recurrencia y el pronóstico, y puede orientar el abordaje. Además, permite la formación de cohortes homogéneas para fines de investigación y futuras intervenciones terapéuticas. Dado que la base genética de SEDh es aún desconocida, el diagnóstico de este subtipo se basa en los hallazgos clínicos, tal como se indica en los criterios revisados para SEDh.
En vista de la gran heterogeneidad genética y la variabilidad fenotípica de los subtipos de SED, y la superposición clínica entre muchos de estos subtipos, pero también con otros TTCs, el diagnóstico definitivo se basa en todos los subtipos, excepto SEDh, en la confirmación molecular con la identificación de la/s variante/s causante/s en el gen respectivo.
Tabla I. Clasificación clínica de los síndromes de Ehlers-Danlos, patrón de herencia y base genética
Píncha en la imagen para verla más grande
Las estrategias de diagnóstico molecular deben basarse en las tecnologías NGS, que ofrecen el potencial para la secuenciación paralela de múltiples genes. La resecuenciación selectiva de un grupo de genes, por ejemplo, COL5A1, COL5A2, COL1A1 y COL1A2, es un enfoque efectivo de tiempo y coste para el diagnóstico molecular del SED genéticamente heterogéneo. Cuando no se identifica mutación (o en caso de una condición autosómica recesiva sólo una mutación), este enfoque debe ser complementado con una estrategia de detección de la variante del número de copias (CNV) para identificar grandes deleciones o duplicaciones, por ejemplo, amplificación de sondas dependientes de ligandos múltiples (MLPA), qPCR, o análisis de matrices dirigidas. Alternativamente, o en una segunda fase, pueden utilizarse las técnicas de secuenciación completa del exoma (WES) o secuenciación del genoma completo (WGS) y secuenciación del ARN, con el análisis de datos enfocándose inicialmente en los genes de interés para un subtipo dado de SED. En ausencia de la identificación de una mutación causante, este enfoque permite ampliar el análisis a otros genes dentro del genoma. Esto es particularmente interesante en vista de la superposición clínica entre los subtipos de SED y con otros TTC y la observación de que en una proporción importante de pacientes con SED no se identifican variantes patógenas en ninguno de los genes conocidos asociados a SED.
La interpretación de variantes de relevancia incierta (VSI), especialmente variantes de cambio de sentido, debe incluir la correlación con el fenotipo clínico completo. De acuerdo con las pautas de ACMG, las variantes que se apoyan en alguna evidencia de patogenicidad (por ejemplo, puntajes altos in silico, presencia en un dominio funcionalmente activo) pueden considerarse «patógenas probables». Los estudios de segregación familiar pueden ayudar a interpretar la patogenicidad de la variante, y para algunos genes, están disponibles análisis de proteínas ultraestructurales, bioquímicas y/o funcionales, como se describe a continuación. Los individuos que albergan una variante «probablemente patógena» deben ser seguidos clínicamente. El asesoramiento inicial para estos pacientes debe señalar que el verdadero significado de la variante no se conocerá hasta que estas pruebas adicionales se hayan completado. A largo plazo, es probable que la asignación de patogenicidad sea facilitada por los datos de los proyectos de secuenciación genómica a gran escala en pacientes y grupos de control [Weerakkody et al., 2016].
Para los pacientes que cumplen el conjunto de requisitos clínicos mínimos para un subtipo específico de SED, pero que no tienen acceso a la confirmación molecular; en el que se identifica uno o más VSI en uno de los genes específicos del subtipo de SED; o en los que no se identifican variantes causantes en ninguno de los genes específicos del subtipo de SED, puede realizarse un «diagnóstico clínico provisional» de un subtipo de SED y los pacientes deben seguirse clínicamente. Sin embargo, se deben considerar los diagnósticos alternativos y, por lo tanto, las pruebas moleculares ampliadas.
MECANISMOS PATOGÉNICOS SUBYACENTES A LOS SÍNDROMES DE EHLERS-DANLOS
Si bien la clasificación propuesta clínicamente tiene como objetivo ser fácil de usar para el no especialista en SED, y ofrece a los pacientes afectados y sus familiares un diagnóstico «descriptivo» que él o ella puedan identificar, una clasificación genética proporciona un mejor marco para la investigación y para el desarrollo de futuras estrategias de tratamiento. Para satisfacer tanto las necesidades clínicas como las de investigación, se propone, además de la clasificación clínica, un esquema patogenético, que agrupa subtipos de SED para los cuales las proteínas, codificadas por los genes causantes, funcionan dentro de la misma vía y que es probable que hayan compartido mecanismos patogénicos, basados en el conocimiento actual (Tabla II). Se propuso un reagrupamiento similar de los subtipos de osteogénesis imperfecta (OI) por función génica y se ha adaptado ampliamente en los contextos clínico y de investigación.
Tabla II. Reagrupación de los síndromes de Ehlers-Danlos de acuerdo a la genética subyacente y a los mecanismos patogénicos
Píncha en la imagen para verla más grande
CLASIFICACIÓN DE LOS SÍNDROMES DE EHLERS-DANLOS
SED clásico (SEDc)
-
- Herencia
Autosómica dominante
-
- Criterios mayores
-
-
-
Hiperextensibilidad de la piel1 y cicatrices atróficas2
-
Hipermovilidad articular generalizada (HAG)3
-
-
-
- Criterios menores
-
-
-
Piel con tendencia a los hematomas 4
-
Piel suave y aterciopelada 5
-
Fragilidad de la piel (o cicatrices traumáticas)
-
Pseudotumores moluscoides 6
-
Esferoides subcutáneos 7
-
Hernia (actual o histórica)
-
Pliegues epicánticos 8
-
Complicaciones de la hipermovilidad articular (por ejemplo, esguinces, luxación /subluxación, dolor, pie plano flexible)
-
Antecedentes familiares de un familiar de primer grado que cumpla con los criterios clínicos
-
-
-
- Criterios mínimos sugerentes para SEDc
– Criterio mayor(1): hiperextensibilidad cutánea y cicatrización atrófica
Más
– Otro criterio mayor (2): Hipermovilidad articular generalizada y/o: al menos tres criterios menores
– La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
-
- Base molecular
Más del 90% de los pacientes con SEDc albergan una mutación heterocigótica en uno de los genes que codifican el colágeno tipo V (COL5A1 y COL5A2) [Symoens et al., 2012; Ritelli et al., 2013; Zoppi et al., 2015] (véase también «Síndrome de Ehlers-Danlos, tipo clásico», de Bowen et al., en este número).
En raras ocasiones, mutaciones específicas en los genes que codifican el colágeno tipo I pueden asociarse con un fenotipo SEDc. Estos incluyen la sustitución heterocigótica COL1A1 c.934C> T, p. (Arg312Cys) [Malfait et al., 2007a]. Los pacientes que albergan esta mutación están particularmente en riesgo de rotura vascular, mientras que los pacientes que albergan otras sustituciones de arginina a cisteina de COL1A1 están asociados con otros fenotipos específicos (véase también «Síndromes de Ehlers-Danlos, Tipos Raros», de Brady et al. ).
Dodecilsulfato sódico y la electroforesis en gel de poliacrilamida (SDS PAGE) demuestra la migración de una banda extra en el fraccionamiento celular, y a veces también en el fraccionamiento medio. Esta banda, que desaparece después de la reducción con ?-mercaptoetanol, consiste en cadenas ? unidas a disulfuro [Malfait et al., 2007b]. Además, las mutaciones bialélicas de COL1A2 que llevan a la ausencia completa de la cadena de colágeno pro?2 (I) también pueden presentar un fenotipo clásico de tipo SED, pero estos pacientes corren el riesgo de desarrollar problemas valvulares cardiacos graves.
Además, la herencia de esta condición es autosómica recesiva (véase también «SED Cardiovalvular», y «Tipos raros del Síndrome de Ehlers Danlos», de Brady et al., En este número). SDS PAGE demuestra la ausencia completa de cadenas (pro) alpha 2 de (pro)colágeno de tipo I extraído de la dermis [Schwarze et al., 2004; Malfait et al., 2006].
-
- Verificación del diagnóstico clínico
Se indica el cribado molecular mediante resecuenciación dirigida de un panel genético que incluye al menos los genes COL5A1, COL5A2, COL1A1 y COL1A2, o por WES o WGS. Cuando no se identifica ninguna mutación, este enfoque debe ser complementado con una estrategia de detección de CNV para identificar grandes deleciones o duplicaciones.
En caso de indisponibilidad de pruebas genéticas, los hallazgos en una microscopía electrónica de transmisión (TEM) de flores de colágeno en la biopsia de piel pueden apoyar el diagnóstico clínico, pero no puede confirmarlo.
La ausencia de estos hallazgos confirmatorios no excluye el diagnóstico, ya que tipos específicos de mutaciones (p. Ej., Mutaciones intrónicas profundas) pueden no ser detectados por técnicas de diagnóstico molecular estándar; Sin embargo, los diagnósticos alternativos deben ser considerados en ausencia de mutaciones COL5A1, COL5A2, COL1A1 o COL1A2.
Más del 90% de los pacientes con SEDC albergan una mutación heterocigótica en uno de los genes que codifican el colágeno tipo V (COL5A1 y COL5A2).
Imagen 1. La piel atrófica / cicatrices ensanchadas visto en SED hipermóvil en comparación con SED clásico

SED Hipermóvil. Cicatriz postraumática, atrófica y ensanchada en un varón joven (A). El estiramiento de la piel entre los dedos del examinador revela una atrofia leve de la dermis subyacente (B). Otra cicatriz atrófica y ensanchada debido al retraso en la cicatrización de la herida después de la extirpación de un nevo melanocítico en una mujer joven (C). SED Clásico. Típica cicatriz papirácea y hemosiderina después de la reapertura repetitiva de la herida y del pseudotumor del moluscoide en un varón adulto (D). Cicatriz papirácea, pero no hemosiderina y laxa en una mujer joven (E). Esferoide subcutáneo (F). Gran pseudotumor moluscoide en el codo (G).
SED clásico II (SEDcl)
-
- Herencia
Autosómica recesiva
-
- Criterios mayores
-
-
-
Hiperextensibilidad de la piel, con textura de piel aterciopelada y ausencia de cicatrices atróficas
-
HAG con o sin luxaciones recurrentes (más comúnmente hombro y tobillo)
-
Piel con tendencia a los hematomas / Equimosis espontánea
-
-
-
- Criterios menores
-
-
-
Deformidades del pie: antepie ancho/regordete, braquidactilia con exceso de piel; pie plano; Hallux valgus; Pápulas piezogénicas.
-
Edema en las piernas en ausencia de insuficiencia cardíaca
-
Debilidad muscular proximal y distal leve
-
Polineuropatía axonal
-
Atrofia de músculos en manos y pies
-
Manos acrogéricas, dedo de mazo, clinodactilia, braquidactilia
-
Prolapso Vaginal/uterino/rectal
-
-
-
- Criterios mínimos sugerentes para los SEDcl
-
-
-
Todos los tres criterios mayores y una historia familiar compatible con la transmisión autosómica recesiva.
-
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
-
-
-
- Base molecular
El SEDcl es causado por una completa falta de Tenascina XB (TNX) debido a las mutaciones bialélicas TNXB, que conducen a una degradación del ARN mensajero mediada por mutaciones terminadoras., o la deleción bialélica de TNXB. Como resultado, la proteína TNX está completamente ausente. TNXB es el único gen asociado con SEDcl.
-
- Verificación del diagnóstico clínico
El análisis molecular del gen TNXB debe utilizarse como prueba estándar de confirmación. Las dificultades en las pruebas de ADN están relacionadas con la presencia de un pseudogen (TNXA), que es más del 97% idéntico al extremo 3 ‘de TNXB (exones 32-44). Con la única excepción del exón 35, que muestra parcialmente una secuencia específica de TNXB, las secuencias de exón y intrón en esta región son idénticas o casi idénticas tanto en el gen como en el pseudogen. Esto tiene implicaciones tanto para el análisis de secuenciación como para el análisis de deleción/duplicación.
Para el análisis de secuencia de TNXB, se recomiendan dos enfoques.
Sanger de todo el gen TNXB.
Secuenciación de próxima generación de la secuencia TNXB + Sanger de la región pseudogénica.
Ambos enfoques requerirán el análisis de secuencia de la región de pseudogen homólogo en unos pocos amplicones multi-exon.
Si no se identifica ninguna o sólo hay una mutación causante por secuenciación clásica, se deben agregar métodos adicionales que permitan la detección de deleciones/duplicaciones grandes. Hasta ahora ningún método es capaz de detectar específicamente CNVs TNXB en los exones altamente homólogos, 32-34 y 36-44. El análisis CNV del exón 35 se usa actualmente para detectar deleciones en esta región, incluyendo la deleción de 30 kb previamente descrita por Schalkwijk et al. [2001].
TNX, una gran glicoproteína de matriz extracelular de 450 kDa, secretada por fibroblastos cutáneos, puede ser detectada con anticuerpos dirigidos contra su extremo carboxiterminal. Los pacientes con SEDcl están completamente exentos de proteína TNX en suero. Se hace referencia al documento de Schalkwijk et al. [2001] para obtener información más detallada sobre el método utilizado para detectar TNX.
La ausencia de estos hallazgos confirmatorios no excluye el diagnóstico, ya que tipos específicos de mutaciones (p. Ej., Mutaciones intrónicas profundas) pueden no ser detectados por técnicas de diagnóstico molecular estándar; sin embargo, los diagnósticos alternativos deben ser considerados en ausencia de una mutación TNXB.
SED cardiovalvular (SEDcv)
-
- Herencia
Autosómica recesiva
-
- Criterios mayores
-
-
-
Problemas progresivos cardíacos-valvulares (válvula aórtica, válvula mitral) 10
-
Afectación cutánea: hiperextensibilidad cutánea, 11 cicatrices atróficas, piel delgada, facilidad para los moretones.
-
-
-
- Criterios menores
-
-
-
Hernia inguinal
-
Deformidad del pectum (especialmente excavatum)
-
Dislocaciones articulares
-
Deformidades de los pies: pies planos, pies planovalgus, hallux valgus
-
-
-
- Criterios mínimos sugerentes para los SEDcv
– Criterio mayor (1): problemas cardíacos-valvulares progresivos severos
– Y una historia familiar compatible con herencia autosómica recesiva
Más
Otro: cualquier otro criterio mayor
y / o: al menos dos criterios menores
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
-
- Base Molecular
SEDcv es causada por una completa falta de la cadena pro?2 de colágeno de tipo I debido a las mutaciones bialélicas COL1A2, que conducen a la Degradación del ARN mensajero mediada por mutaciones terminadoras. COL1A2 es el único gen asociado con SEDcv.
-
- Verificación del diagnóstico
Se indica el cribado molecular por secuenciación de Sanger de COL1A2, o la resecuenciación dirigida de un panel de genes que incluye COL1A2. Cuando no se identifica ninguna mutación, este enfoque debe ser complementado con una estrategia de detección de CNV para identificar grandes deleciones o duplicaciones.
En caso de no disponer de pruebas genéticas, SDS PAGE demuestra la ausencia total de cadenas de colágeno (pro) ?2 (I).
Mientras que la ausencia de estos hallazgos bioquímicos confirmatorios permite excluir el diagnóstico de SEDcv, la ausencia de estos hallazgos genéticos confirmatorios no excluye el diagnóstico, ya que tipos específicos de mutaciones (por ejemplo, mutaciones intrónicas profundas) pueden no ser detectados por técnicas moleculares de diagnóstico estándar.
SED vascular (SEDv)
- Herencia
Autosómica dominante
- Criterios mayores
-
Antecedentes familiares de SEDv con una variante causante documentada en COL3A1
-
Ruptura arterial a una edad temprana
-
Perforación espontánea del colon sigmoideo en ausencia de enfermedad diverticular conocida u otra patología intestinal
-
Ruptura uterina durante el tercer trimestre en ausencia cesáreas previas y/o desgarros perineales anteriores al parto.
-
Formación de fístula del seno carotídeo cavernoso (CCSF) en ausencia de trauma
- Criterios menores
-
Moretones no relacionados con un trauma identificado y/o en sitios inusuales como las mejillas y la espalda
-
Piel delgada, translúcida, con visibilidad venosa incrementada
-
Apariencia facial característica
-
Neumotórax espontáneo
-
Acrogeria
-
Pies equinovarus
-
Dislocación congénita de cadera
-
Hipermovilidad de pequeñas articulaciones
-
Rotura muscular y tendinosa
-
Queratocono
-
Recesión gingival y fragilidad gingival
-
Venas varicosas de inicio temprano (menores de 30 años y nulíparas en mujeres)
- Criterios mínimos sugerentes para los SEDv
Una historia familiar del trastorno, ruptura o disección arterial en individuos menores de 40 años de edad, ruptura inexplicada de colon sigmoideo o neumotórax espontáneo en presencia de otras características consistentes con SEDv deben conducir a estudios diagnósticos para determinar si el individuo tiene SEDv . Las pruebas para SEDv también deben considerarse en presencia de una combinación de las otras características clínicas «menores» enumeradas anteriormente.
Incluso para los clínicos experimentados el diagnóstico clínico de SEDv puede ser difícil. Debido a las implicaciones para el tratamiento, la historia natural y el riesgo de recurrencia, el diagnóstico de SEDv se basa en la identificación de una variante causante en un alelo de COL3A1.
- Base Molecular
Los pacientes con SEDv típicamente albergan una mutación heterocigótica en el gen COL3A1, que codifica el colágeno tipo III, con la rara excepción de las mutaciones heterozigóticas específicas de sustitución de arginina a cisteína en COL1A1 (c.934C> T, p.Arg312Cys; c.1720C> T , P.Arg574Cys y c.3277C> T, p.Arg1093Cys) que también están asociadas con fragilidad vascular, mimetizando COL3A1-SEDv [Malfait et al., 2007b], (véase también «Tipo raros de Síndromes de Ehlers Danlos», por Brady et al., en este número).
En casos muy raros, se pueden identificar variantes patógenas bialélicas en COL3A1.
- Verificación del diagnóstico clínico
Se indica la selección molecular por secuenciación de Sanger de COL3A1, o resecuenciación dirigida de un panel de genes que incluye COL3A1 y COL1A1 (este último para identificar las mutaciones de sustitución de arginina a cisteína antes mencionadas). Cuando no se identifica ninguna mutación, este enfoque debe ser complementado con una estrategia de detección de CNV para identificar grandes deleciones o duplicaciones.
La ausencia de estos hallazgos confirmatorios no excluye el diagnóstico, ya que tipos específicos de mutaciones (p. Ej., Mutaciones intrónicas profundas) pueden no ser detectados por técnicas de diagnóstico molecular estándar; Sin embargo, los diagnósticos alternativos deben ser considerados en ausencia de una mutación COL3A1 o COL1A1.
“Una historia familiar del trastorno, ruptura o disección arterial en individuos menores de 40 años de edad, ruptura inexplicada de colon sigmoideo o neumotórax espontáneo en presencia de otras características consistentes con SEDv deben conducir a estudios diagnósticos para determinar si el individuo tiene SEDv .”
SED hipermóvil (SEDh)
- Herencia
Autosómica dominante
- Base Molecular
Desconocida
- Diagnóstico clínico
El diagnóstico de SEDh sigue siendo clínico, ya que aún no existe una etiología genética fiable o apreciable que se pueda realizar en la gran mayoría de los pacientes. Esto, en parte, probablemente refleja la heterogeneidad genética. Además, la presentación sindrómica puede variar según la edad y el género. También existe un espectro clínico que va desde la hipermovilidad articular asintomática, hasta la hipermovilidad «no sindrómica» con manifestaciones secundarias, hasta el SEDh (ver «Marco para la clasificación de la hipermovilidad articular y condiciones relacionadas» de Castori et al, en este número). Un diagnóstico de SEDh debe ser asignado sólo en aquellos que cumplan con todos los criterios descritos a continuación, lo que debería ayudar a reducir la heterogeneidad y facilitar los esfuerzos para descubrir la/s causa/s genética/s subyacente del síndrome que, a su vez, puede ayudar al abordaje clínico. Debido a que actualmente no existe una prueba de laboratorio «estándar de oro» para confirmar o refutar el diagnóstico, anticipamos que la investigación futura llevará a revisiones adicionales de estos criterios clínicos que requieren una revisión regular de la literatura médica pertinente. También es imprescindible, como se trata de un diagnóstico clínico, estar relativamente seguro de que la presentación del paciente no representa uno de los muchos otros trastornos del tejido conectivo. Por lo tanto, el clínico debe ser experimentado en el examen físico descrito aquí, así como la presentación histórica y clínica de otros TTC y sus diagnósticos.
El diagnóstico clínico de SEDh necesita la presencia simultánea de los criterios 1 Y 2 Y 3.
Anotaciones específicas y explicaciones adicionales (es decir, notas de pie de página [FN]) se informan para las características seleccionadas.
- Criterio 1: Hipermovilidad Articular Generalizada (HAG)
Hasta la fecha, la puntuación de Beighton (Figura 2) es la herramienta más reconocida para evaluar HAG (ver «Propiedades de Medición de los Métodos de Evaluación Clínica para Clasificar la Hipermovilidad Conjunta Generalizada -una Revisión Sistemática» de Juul-Kristensen et al.en este número). De acuerdo con la definición original de la puntuación de Beighton y su posterior incorporación en la nosología de Villefranche para los SEDh, el corte para la definición de HAG es ?5 puntos sobre 9. Sin embargo, el rango de movimiento conjunto disminuye con la edad [Soucie et Al., 2011; McKay et al., 2016] y existe una relación inversa entre la edad en la determinación y la puntuación de Beighton [Remvig et al., 2007], por lo que el corte de cinco puede provocar un sobrediagnóstico en los niños y un subdiagnóstico entre adultos y ancianos. Como HAG se considera un requisito previo para el diagnóstico de SEDh y la HAF es un rasgo constitucional fuertemente influenciado por condiciones adquiridas y heredadas (por ejemplo, sexo, edad, traumas pasados, comorbilidades, etc.), algunas pequeñas adaptaciones deben ser consideradas para el diagnóstico de SEDh. El Comité, en nombre del Consorcio Internacional sobre los Síndromes de Ehlers-Danlos, propone ?6 para los niños prepúberes y adolescentes, ?5 para los hombres y mujeres púberes hasta los 50 años, y ?4 para los> 50 años de edad para SEDh. Esto puede variar de otros tipos de SED, pero estos tipos tienen pruebas confirmatorias.
De acuerdo con la definición original de la puntuación de Beighton y su posterior incorporación en la nosología de Villefranche para los SEDh, el corte para la definición de HAG es ?5 puntos sobre 9. Sin embargo, el rango de movimiento conjunto disminuye con la edad [Soucie et Al., 2011; McKay et al., 2016] y existe una relación inversa entre la edad en la determinación y la puntuación de Beighton [Remvig et al., 2007], por lo que el corte de cinco puede provocar un sobrediagnóstico en los niños y un subdiagnóstico entre adultos y ancianos..
En los individuos con limitaciones articulares adquiridas (cirugía pasada, silla de ruedas, amputaciones, etc.) que afectan al cálculo de la puntuación de Beighton, la evaluación de HAG puede incluir información histórica utilizando el cuestionario de cinco puntos (Cuadro III) [Hakim y Grahame, 2003 ; Mulvey et al., 2013], aunque esto no ha sido validado en niños (ver «Propiedades de Medición de los Métodos de Evaluación Clínica para Clasificar la Hipermovilidad Conjunta Generalizada -una Revisión Sistemática» de Juul-Kristensen et al. Si la puntuación de Beighton es 1 punto por debajo del límite de edad y sexo específico y el 5PQ es «positivo» (= al menos dos puntos positivos), entonces se puede hacer un diagnóstico de HAG.
Tabla III. El Cuestionario de Cinco Puntos. Adaptado De [Grahame y Hakim, 2003]
1. ¿Puede usted ahora (o podría usted alguna vez) poner sus manos llanas en el suelo sin doblar sus rodillas?
2. ¿Puede usted ahora (o podría usted alguna vez) doblar su pulgar para tocar su antebrazo?
3. Cuando era niño, ¿divirtió a sus amigos contorsionando su cuerpo en formas extrañas o podría hacer las divisiones?
4. ¿Como niño o adolescente, su hombro o rodilla se dislocaron en más de una ocasión?
5. ¿Consideras que tienes» articulaciones dobles»?
Una respuesta «sí» a dos o más preguntas sugiere hipermovilidad articular con 80-85% de sensibilidad y 80-90% de especificidad
Figura 2. El sistema de puntuación de Beighton
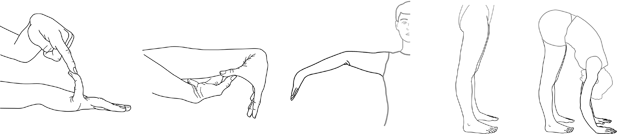
Cada articulación se mide usando un goniómetro y cada lado se califica de forma independiente como se ha descrito [Juul-Kristensen et al., 2007]. (A) Con la palma de la mano y el antebrazo apoyados sobre una superficie plana con el codo flexionado a 90 °, si la articulación metacarpo-falángica del quinto dedo puede ser hiperextendida más de 90 ° con respecto al dorso de la mano, Se considera positivo, puntuando 1 punto. (B) Con los brazos extendidos hacia adelante pero con pronación manual, si el pulgar puede ser pasivamente movido para tocar el antebrazo ipsilateral, se considera positivo puntuando 1 punto. (C) Con los brazos extendidos hacia un lado y la mano en decúbito supino, si el codo se extiende más de 10 °, se considera positivo anotando 1 punto. (D) Mientras está de pie, con las rodillas bloqueadas en el genu recurvatum, si la rodilla se extiende más de 10 °, se considera positiva la puntuación de 1 punto. (E) Con las rodillas bloqueadas y los pies juntos, si el paciente puede doblar hacia adelante para colocar la palma total de ambas manos en el suelo justo delante de los pies, se considera positiva puntuación 1 punto. La puntuación total es 9. Figura cortesía del Dr. Juul-Kirstensen.
Para los pacientes con puntuaciones más bajas de Beighton, a menudo se tiene en consideración la evaluación de otras articulaciones, incluyendo articulación temporomandibular, hombro, cadera, pie, muñeca, tobillo y otros dígitos. La flexión del tobillo y dorsiflexión de la muñeca aumentadas, la rotación aumentada interna y externa de la cadera y el pie plano se han correlacionado con la puntuación de Beighton [Smits-Engelsman et al., 2011] Sin embargo, preocupaciones similares sobre edad, género e influencias ambientales,así como la metodología de medición y valores de corte fiables, limitan este análisis y lo hacen demasiado subjetivo en la determinación de HAG. Por lo tanto, el uso de tales mediciones no puede ser factorizado en un algoritmo de diagnóstico en este momento. Obviamente, se necesita más información acerca de la(s) metodología(s) de evaluación en la determinación de HAG (ver «Propiedades de Medición de los Métodos de Evaluación Clínica para Clasificar la Hipermovilidad Conjunta Generalizada -una Revisión Sistemática» por Juul-Kristensen y otros, en este número).
Por último, el uso del sistema de puntuación Beighton está destinado a ser un método de detección diagnóstica. Se entiende que el género, la edad, el origen étnico, el entrenamiento de fuerza, los ejercicios de estiramiento y el calentamiento afectan a HA y por lo tanto a HAG. Sin embargo, la sobrecompensación muscular, las lesiones y la cirugía pueden causar hipermovilidad o hipomobilidad articular. La sobrecompensación muscular, como los isquiotibiales tensos, puede afectar negativamente el grado de extensión de la rodilla y la flexión lumbar, mientras que los ejercicios de estiramiento y calentamiento afectan positivamente. Las lesiones pueden desestabilizar una articulación o, alternativamente, reducir el movimiento. La cirugía también puede afectar a una articulación. Por ejemplo, una persona con fusión de la columna lumbar puede no ser capaz de tener una flexión espinal «positiva» hacia adelante para la puntuación de Beighton. Hay intención de los médicos de considerar esto una puntuación positiva, pero sin capacidad actual o demostración histórica, se debe anotar negativo. Un argumento podría ser hecho para invalidar la flexión de la columna vertebral por lo que la puntuación total sería de ocho y no nueve. Sin embargo, no se sabe si el numerador (determinante de HAG) debe ajustarse en esta situación. En teoría, esto tiene sentido, pero ¿cuál es el límite apropiado? Por lo tanto, al igual que cualquier herramienta clínica, hay cierta subjetividad y esta no es una guía para reemplazar el juicio del clínico experimentado; Ssi embargo, se requiere la estandarización de los procedimientos de desempeño. Es posible que se desee etiquetar a estas personas como «probable HAG», pero en la actualidad, «probable HAG» no debe considerarse una alternativa de la HAG objetivamente diagnosticada (como se describe más arriba) en el diagrama de flujo de diagnóstico de SEDh. Debería contemplarse un examen más estricto de las fenocopias.
-
Criterio 2: Dos o más de las siguientes características (A-C) DEBEN estar presentes (por ejemplo: A y B, A y C, B y C, A y B y C)
– Característica A: manifestaciones sistémicas de un trastorno de tejido conectivo más generalizado (un total de cinco deben estar presentes) 12
-
Piel anormalmente suave o aterciopelada 13
-
Hiperextensibilidad de la piel leve14
-
Estrías inexplicadas como estrías distensas o rubor en la espalda, las ingles, los muslos, los pechos y/o el abdomen en adolescentes, hombres o mujeres prepúberes sin antecedentes de aumento significativo o pérdida de grasa o peso corporal
-
Pápulas piezógenas bilaterales del talón 15
-
Hernia abdominal recurrente o múltiple (p. Ej., Umbilical, inguinal, crural)
-
Cicatrización atópica que involucra al menos dos sitios y sin la formación de verdaderas cicatrices papiráceas y/o hemosiderinas como se observa en el SED clásico 16
-
Prolapso del suelo pélvico, rectal y/o uterino en niños, hombres o mujeres nulíparas sin antecedentes de obesidad mórbida u otra condición médica predisponente conocida
-
El hacinamiento dental y paladar alto o estrecho 17
-
Aracnodactilia, según lo definido en uno o más de lo siguiente: signo positivo de la muñeca (signo de Steinberg) en ambos lados; signo de pulgar positivo (signo de Walker) en ambos lados
-
Alcance del brazo a la altura ?1.05
-
Prolapso de la válvula mitral (MVP) leve o mayor basado en estrictos criterios ecocardiográficos 18
-
Dilatación de la raíz aórtica con puntuación Z> +2
– Característica B: antecedentes familiares positivos, con uno o más familiares de primer grado que cumplen de forma independiente los criterios diagnósticos actuales para SEDh
– Característica C: complicaciones músculo-esqueléticas (debe tener al menos una)
-
Dolor musculoesquelético en dos o más extremidades, que se repite diariamente durante al menos 3 meses
-
Dolor crónico, extenso para ?3 meses
-
Dislocaciones articulares recurrentes o inestabilidad franca de las articulaciones, en ausencia de trauma (a o b) 19
-
-
Tres o más luxaciones atraumáticas en la misma articulación o dos o más luxaciones atraumáticas en dos articulaciones diferentes que ocurren en diferentes momentos
-
Confirmación médica de inestabilidad articular en dos o más sitios no relacionados con traumatismos 20
-
Criterio 3: Todos los siguientes requisitos deben ser cumplidos
Ausencia de la fragilidad inusual de la piel, que debe considerar de otros tipos de SED
Exclusión de otros trastornos del tejido conectivo hereditarios y adquiridos, incluyendo condiciones reumatológicas auto-inmunes. En los pacientes con un trastorno del tejido conectivo adquirido (por ejemplo, lupus, artritis reumatoide, etc.), el diagnóstico adicional de SEDh requiere reunir tanto las características A como B del Criterio 2. La característica C del Criterio 2 (dolor crónico e inestabilidad) no puede ser contado para un diagnóstico de SEDh en esta situación.
Exclusión de diagnósticos alternativos que también pueden incluir hipermovilidad articular mediante hipotonía y/o laxitud de tejido conectivo. Los diagnósticos alternativos y las categorías diagnósticas incluyen, pero no se limitan a, trastornos neuromusculares (por ejemplo, SED miopático, miopatía de Bethlem), otros TTC (por ejemplo, otros tipos de SED, síndrome de Loeys-Dietz, síndrome de Marfan) y displasias esqueléticas OI). La exclusión de estas consideraciones puede basarse en antecedentes, examen físico y/o pruebas genéticas moleculares, como se indica.
Comentario general
Muchas otras características se describen en SEDh, pero la mayoría no son lo suficientemente específicos ni sensibles por el momento para ser incluidos en los criterios de diagnóstico formal (véase «Síndrome de Ehlers-Danlos Hipermovil (también conocido como síndrome de Ehlers-Danlos Tipo III y síndrome de Ehlers-Danlos tipo hipermobilidad) Descripción clínica e historia natural «de Tinkle et al., en este número). Estos incluyen pero no se limitan a: trastornos del sueño, fatiga, taquicardia ortostática postural, trastornos gastrointestinales funcionales, disautonomía, ansiedad y depresión. Estas otras manifestaciones sistémicas pueden ser más debilitantes que los síntomas de las articulaciones, a menudo deterioran la funcionalidad y la calidad de vida y siempre deben determinarse durante los encuentros clínicos. Aunque no forman parte de los criterios diagnósticos, la presencia de tales manifestaciones sistémicas puede dar lugar a la consideración de los SEDh en el diagnóstico diferencial. La investigación futura deberá enfocarse en tales síntomas para validar cualquier asociación con SEDh, describir subgrupos o subfenotipos y centrarse en el manejo basado en la evidencia de los síntomas en el contexto de SEDh.
SED artrocalacsia (SEDa)
- Herencia
Autosómico dominante
- Criterios mayores
-
Dislocación bilateral de cadera congénita 21
-
HAG grave, con múltiples luxaciones/subluxaciones 22
-
Hiperextensibilidad de la piel 22
- Criterios menores
- Hipotonía muscular
- Cifoscoliosis
- Osteopenia radiológicamente leve
- Fragilidad del tejido, incluyendo cicatrices atróficas
- Piel con tendencia a los hematomas
- Criterios mínimos sugerentes para SEDa:
– Criterio mayor (1): Dislocación congénita bilateral de cadera
Más
– Otro Criterio mayor (3): hiperextensibilidad de la piel
– Otro Criterio mayor (2): HAG grave con múltiples luxaciones / subluxaciones y al menos otros dos criterios menores
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
- Base Molecular
El SEDa es causado por mutaciones heterocigotas en cualquiera de COL1A1 o COL1A2, que causan pérdida total o parcial del exón 6 del gen respectivo. No hay otros genes asociados con SEDa.
- Verificación del diagnóstico
Está indicada la selección molecular por secuenciación de Sanger de COL1A1 y COL1A2, o resecuenciación dirigida de un panel genético que incluye estos genes. Cuando no se identifica ninguna mutación, este enfoque debe ser complementado con una estrategia de detección de CNV para identificar grandes deleciones o duplicaciones.
En el caso de indisponibilidad de pruebas genéticas, el SDS PAGE del colágeno soluble en pepsina en el medio o capa celular de fibroblastos dérmicos cultivados demuestra la presencia de una cadena mutante pN?1 (I) o pN?2 (I) (cadenas de procolágeno precursoras en las que el carboxi (C) -pero no el amino (N)- propetido está escindido.
El TEM de especímenes de piel muestra fibrillas de colágeno organizadas de forma aleatoria con un diámetro más pequeño y más variable, y un contorno irregular. Estos hallazgos pueden apoyar el diagnóstico, pero no pueden confirmarlo.
La ausencia de una mutación causante en COL1A1 o COL1A2 que conduce a la deleción completa o parcial del exón 6 de cualquiera de los genes excluye el diagnóstico de SEDa.
SED dermatosparaxis (SEDd)
- Herencia
Autosómica recesiva
- Criterios mayores
-
Fragilidad extrema de la piel con rotura de piel congénita o postnatal
-
Rasgos craneofaciales, que son evidentes al nacer o en la primera infancia, o evolucionan más tarde en la infancia23
-
Piel redundante, casi laxa, con excesivos pliegues cutáneos en las muñecas y los tobillos
-
Aumento de arrugas palmar
-
Severa propensión a hematomas con riesgo de hematomas subcutáneos y hemorragia
-
Hernia umbilical
-
Retraso del crecimiento postnatal
-
Miembros cortos, manos y pies
-
Complicaciones perinatales debidas a la fragilidad del tejido conectivo 24
- Criterios menores
-
Textura suave y pastosa de la piel
-
Hiperextensibilidad de la piel
-
Cicatrices atróficas
-
HAG 25
-
Las complicaciones de la fragilidad visceral (por ejemplo, ruptura de la vejiga, rotura diafragmática, prolapso rectal)
-
Retraso del desarrollo motor 26
-
Osteopenia
-
Hirsutismo
-
Anomalías dentales
-
Errores refractivos (miopía, astigmatismo)
-
Estrabismo
- Criterios mínimos sugerentes para SEDd
– Criterio mayor (1): fragilidad extrema de la piel
– Y criterio mayor (2): rasgos craneofaciales características
Más:
– otro criterio mayor
– Y / o: tres criterios menores
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
- Base molecular
El SEDd es causado por mutaciones bialélicas en ADAMTS2, el gen que codifica ADAMTS-2, el principal procollageno I N-proteinasa. Es el único gen asociado con SEDd.
- Verificación del diagnóstico
Se indica la selección molecular por secuenciación de Sanger de resecuenciación dirigida de un panel de genes que incluye ADAMTS2. Cuando no se identifica, o sólo una mutación causante, este enfoque debe complementarse con una estrategia de detección de CNV para identificar grandes deleciones o duplicaciones.
En caso de indisponibilidad de pruebas genéticas, el SDS, PAGE demuestra la presencia de cadenas pN?1 (I) y pN?2 (I) de procolágeno tipo I extraído de la dermis en presencia de inhibidores de proteasa o detectado en cultivos de fibroblastos.
El TEM muestra fibrillas de colágeno en muestras de piel afectadas con un patrón jeroglífico. Estos hallazgos ultraestructurales suelen ser típicos, pero pueden ser casi indistinguibles de los observados en el SEDa. Como tal, no son suficientes para confirmar el diagnóstico.
La ausencia de estos hallazgos confirmatorios no excluye el diagnóstico de SEDd, ya que tipos específicos de mutaciones (p. Ej., Mutaciones intrónicas profundas) pueden no ser detectados por las técnicas de diagnóstico mlecular estándar; Sin embargo, los diagnósticos alternativos deben ser considerados en ausencia de mutaciones de ADAMTS2.
SED cifoscoliótico (SEDk)
- Herencia
Autosómica recesiva
- Criterios mayores
-
-
Hipotonía congénita del músculo 27
-
Cifoescoliosis congénita o de inicio precoz (progresiva o no progresiva) 28
-
HAG 29 con luxaciones/subluxaciones (hombros, caderas y rodillas en particular)
-
- Criterios menores
-
Hiperextensibilidad de la piel 29
-
Propensión a los hematomas
-
Ruptura / aneurisma de una arteria de tamaño medio
-
Osteopenia/osteoporosis
-
Escleróticas azules
-
Hernia (umbilical o inguinal)
-
Deformidad del pectum
-
Habito Marfanoide
-
Pies equinovarus
-
Errores de refracción (miopía, hipermetropía)
- Criterios menores específicos de genes
–PLOD1
- – Fragilidad de la piel (propensión a los hematomas, piel friable, cicatrización pobre de heridas), cicatrices atróficas ensanchadas
- – Fragilidad/ruptura escleral y ocular 30
- – Microcornea
- – Dismorfología facial 31
–FKBP14
- – Fragilidad de la piel (propensión a los hematomas, piel friable, cicatrización pobre de heridas), cicatrices atróficas ensanchadas
- – Fragilidad/ruptura escleral y ocular 30
- – Microcornea
- – Dismorfología facial 31
- Criterios mínimos sugerentes para SEDk
– El Criterio mayor (1): hipotonía congénita del músculo
– Y el criterio mayor (2): la cifoscoliosis congénita o de inicio precoz
Más
– Criterio mayor (3): HAG
– Y/o tres criterios menores (ya sea generales o específicos de genes específicos)
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
- Base Molecular
La mayoría de los pacientes con cifoescoliosis alberga mutaciones bialélicas en PLOD1, el gen que codifica la enzima modificadora del colágeno procolageno-lisina, 2-oxoglutarato 5-dioxigenasa 1 (PLOD1 o LH1 [lisilhidroxilasa1]). LH1 juega un papel importante como enzima modificadora postraduccional en la biosíntesis de colágeno mediante hidroxilación de residuos lisilo helicoidales en secuencias de colágeno-Xaa-Lys-Gly a residuos hidroxi-lisilo que sirven como sitios de unión para unidades de carbohidratos (galactosa o glucosil-galactosa) y en la formación de enlaces cruzados intra e intermoleculares de colágeno. La deficiencia de LH1 da como resultado la subhidroxilación de residuos de lisilo y la sub-glicosilación de residuos de hidroxilsiilo en colágenos y, por lo tanto, alteración de la formación de enlaces cruzados con la consecuente inestabilidad mecánica de los tejidos afectados.
Recientemente, se han identificado mutaciones bialélicas en FKBP14, que codifica FKBP22, un miembro de la familia de unión a F506 de las isomerasas cis-trans de peptidil-prolilo, en pacientes que presentan un fenotipo que se superpone clínicamente con kEDS-PLOD1 [Baumann et al., 2012 ].
- Verificación del diagnóstico
La confirmación en laboratorio del SEDk debe comenzar con la cuantificación de enlaces cruzados de desoxipiridinolina (Dpyr o LP para lisil-piridinolina) y piridinolina (Pyr o HP para hidroxilil-piridinolina) en orina mediante cromatografía líquida de alto rendimiento (HPLC). Un aumento del ratio Dpyr/Pyr es una prueba altamente sensible y específica para SEDk causado por mutaciones bialélicas PLOD1 (SEDk-PLOD1), pero es normal para mutaciones bialelicas FKBP14 (kEDS-FKBP14).
El ratio normal de enlaces cruzados Dpyr/Pyr es de aproximadamente 0,2, mientras que en SEDk-PLOD1 la relación aumenta significativamente (aproximadamente 10-40 veces el aumento, rango 2-9). Este método es rápido y rentable y también puede usarse para determinar el estado patogénico de una VUS en PLOD1.
El SDS-PAGE puede detectar una migración más rápida de las cadenas de colágeno subhidroxiladas y sus derivados en SEDk-PLOD1 pero no en SEDk-FKBP14. Sin embargo, las anomalías en la migración pueden ser sutiles.
El análisis molecular de SEDk-PLOD1 puede comenzar con el análisis MLPA de PLOD1, para la evaluación de la duplicación intragénica común en PLOD1 causada por una recombinación Alu-Alu entre los intrones 9 y 16 (el alelo mutante más común) [Hautala et al., 1993 ].
La detección molecular mediante resecuenciación dirigida de un panel de genes que incluye PLOD1 y FKBP14, está indicada cuando MLPA de PLOD1 no logra identificar la duplicación común. Dicho panel genético también incluye otros genes asociados con fenotipos que se superponen clínicamente con SEDk, tales como ZNF469, PRDM5, B4GALT7, B3GALT6, SLC39A13, CHST14 y DSE. Alternativamente, el WES puede ser realizado. Cuando ninguna, o sólo una mutación causante se identifica, este enfoque debe complementarse con una estrategia de detección de CNV para identificar grandes deleciones o duplicaciones en estos genes.
El TEM en muestras de piel ha mostrado diámetros variables y contornos anormales de las fibrillas de colágeno y espacio interfibrilar irregular, pero estas anomalías no son exclusivas de esta condición. Como tal, mientras que el TEM en una biopsia de piel puede apoyar el diagnóstico, no puede confirmarlo.
Mientras que la ausencia de una relación urinaria anormal de LP / HP excluye el diagnóstico de cifoescoliotico-PLOD1, la ausencia de los hallazgos genéticos confirmatorios no excluye el diagnóstico de SEDk, ya que los tipos específicos de mutaciones (por ejemplo, mutaciones intrónicas profundas) pueden no ser detectados por las técnicas de diagnóstico molecular estándar y/u otros genes aún no descubiertos pueden estar asociados con este fenotipo; Sin embargo, los diagnósticos alternativos deben considerarse en ausencia de mutaciones PLOD1 o FKBP14.
La mayoría de los pacientes con cifoescoliosis alberga mutaciones bialélicas en PLOD1, el gen que codifica la enzima modificadora del colágeno procolageno-lisina, 2-oxoglutarato 5-dioxigenasa 1 (PLOD1 o LH1 [lisilhidroxilasa1])..
Imagen 3. Pápulas piezogénicas podálicas
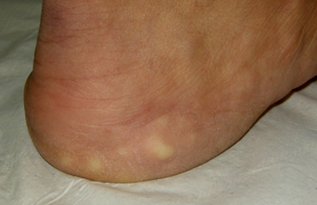
Papulas piezogenicas de los pies que son herniaciones de grasa subcutánea a través de la fascia. A menudo aparecen como nódulos blancos sólo mientras se está cargando peso.
Síndrome de Córnea Frágil (SCF)
- Herencia
Autosómica recesiva
- Criterios mayores
-
Córnea fina, con o sin ruptura (grosor central de la córnea a menudo <400 ?m)
-
Queratocono progresivo de comienzo temprano
-
Queratoglobo progresivo de inicio temprano
-
Escleróticas azules
- Criterios menores
-
Enucleación o cicatrización corneal como resultado de ruptura previa
-
Pérdida progresiva de la profundidad del estroma corneal, especialmente en la córnea central
-
Miopía alta, con longitud axial normal o moderadamente aumentada
-
Desprendimiento de retina
-
Sordera, a menudo con componentes conductivos y sensorioneurales mixtos, frecuencias progresivas, frecuencias más frecuentemente afectadas más severamente (audiograma de tono puro «inclinado»),
-
Membranas timpánicas hiperreceptivas
-
Displasia del desarrollo de la cadera
-
Hipotonía en la infancia, generalmente leve si está presente
-
Escoliosis
-
Aracnodactilia
-
Hipermovilidad de las articulaciones distales
-
Pies planos, hallux valgus
-
Contracciones suaves de los dedos (especialmente el quinto)
-
Piel suave y aterciopelada, piel translúcida
- Criterios mínimos sugerentes para SCF
– Criterio mayor (1): córnea fina, con o sin ruptura (corteza corneal central a menudo <100 micrómetros)
Más
– Al menos otro criterio mayor
– Y/o otros tres criterios menores
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final
- Base Molecular
El BCS es causado por mutaciones bialélicas en ZNF469, que codifican ZNF469, una proteína de dedos de cinc de función desconocida, o PRDM5, que codifica un factor de transcripción de unión a ADN de la familia de proteínas PR / SET que carece de la actividad de la histona metiltransferasa intrínseca. Al menos una familia con un fenotipo BCS clínico no alberga mutaciones en estos genes, lo que sugiere que al menos otro gen podría estar asociado con BCS [Rohrbach et al., 2013].
- Verificación del diagnóstico
Se indica el cribado molecular mediante resecuenciación dirigida de un panel genético que incluye ZNF469 y PRDM5. Dicho panel genético también incluye otros genes asociados con fenotipos que se superponen clínicamente con BCS, tales como PLOD1, FKBP14, B4GALT7, B3GALT6, SLC39A13, CHST14 y DSE. Alternativamente, el WES puede ser realizado. Cuando ninguna, o sólo una mutación causante se identifica, este enfoque debe complementarse con una estrategia de detección de CNV para identificar grandes deleciones o duplicaciones en estos genes.
La ausencia de estos hallazgos confirmatorios no excluye el diagnóstico, ya que tipos específicos de mutaciones (p. Ej., Mutaciones intrónicas profundas) pueden pasar desapercibidos por las técnicas moleculares de diagnóstico estándar, y otros genes aún desconocidos podrían estar asociados con BCS.
SED espondilodisplasico (SEDsp)
- Herencia
Autosómica recesiva
- Criterios mayores
-
Estatura baja (progresiva en la infancia)
-
La hipotonía muscular (que va desde una congénita grave hasta una aparición tardía leve)
-
Inclinación de los miembros
- Criterios menores
-
Hiperextensibilidad de la piel, 32 piel suave y pastosa, piel translúcida delgada
-
Pie plano
-
Desarrollo motor retardado
-
Osteopenia
-
Retraso en el desarrollo cognitivo
- Criterios menores específicos de genes
– B4GALT7
- – Sinostosis radioulnar
- – Contracción del codo bilateral o movimiento limitado del codo
- – HAG 32
- – Un único pliegue palmar transversal
- – Rasgos craneofaciales 33
- – Hallazgos radiográficos característicos 34
- – Hipermetropía severa
- – Córnea encerada
– B3GALT6
- – Cifoescoliosis (congénita o de inicio temprano, progresiva)
- – Hipermovilidad articular, generalizada o restringida a las articulaciones distales, con dislocaciones articulares
- – Contracturas articulares (congénitas o progresivas) (especialmente manos)
- – Dedos peculiares (delgados, cónicos, aracnodactilia, con falanges distales grandes)
- – Pies equinovaros
- – Rasgos craneofaciales característicos 35
- – Decoloración de los dientes, dientes displásicos
- – Hallazgos radiográficos característicos 36
- – Osteoporosis con múltiples fracturas espontáneas
- – Aneurisma aórtico ascendente
- – Hipoplasia pulmonar, enfermedad pulmonar restrictiva
– SLC39A13
- – Ojos protuberantes con escleróticas azuladas
- – Manos con palmas finamente arrugadas
- – Atrofia de los músculos tenares y dedos afilados
- – Hipermovilidad de las articulaciones distales
- – Hallazgos radiológicos característicos 37
- Criterios mínimos sugerentes para los SEDsp
Criterio mayor (1): baja estatura
– Y Criterio mayor (2): hipotonía muscular
Más
– Anomalías radiográficas típicas y al menos otros tres criterios menores (generales o específicos del tipo)
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
- Base Molecular
El SEDsp es causado por:
Mutaciones bialélicas en B4GALT7, que codifican la galactosiltransferasa I (?1,4-galactosiltransferasa 7 o ?4GalT7), que cataliza la transferencia de la primera galactosa al residuo de xilosa en la región de enlace tetrasacárido de glicosaminoglicanos (GAGs).
Mutaciones bialélicas en B3GALT6, que codifican la galactosiltransferasa II (?1,3-galactosiltransferasa 6 o ?3GalT6), que cataliza la transferencia de la segunda galactosa al primer residuo de galactosa en la región de unión del tetrasacárido de GAGs.
Mutaciones bialélicas en SLC39A13, que codifican la proteína homodimérica transmembrana Zrt / irt-like 13 (ZIP13), un miembro de la familia SLC39A / ZIP que regula la afluencia de Zn en el citosol.
- Verificación del diagnóstico
Se indica el cribado molecular mediante resecuenciación dirigida de un panel genético que incluye B4GALT7, B3GALT6 y SLC39A13. Dicho panel genético también incluye otros genes asociados con fenotipos que se superponen clínicamente con SEDsp, tales como PLOD1, FKBP14, ZNF469, PRDM5, CHST14 y DSE. Alternativamente, el WES puede ser realizado. Cuando ninguna, o sólo una, mutación causante se identifica, este enfoque debe complementarse con una estrategia de detección de CNV para identificar grandes deleciones o duplicaciones en estos genes.
Para la prueba definitiva de la deficiencia de GAG ??(mutaciones de B4GALT7 y de B3GALT6), los métodos bioquímicos para evaluar la síntesis de GAG ??en los fibroblastos cultivados de los pacientes están disponibles actualmente en muchos laboratorios especializados [Talhaoui et al., 2010].
La medición en laboratorio de piridinolinas urinarias, lisil-piridinolina (LP) e hidroxilisil-piridinolina (HP) cuantificadas por HPLC permite la detección de un ratio LP/HP aumentado a aproximadamente 1 (comparada con valores normales de aproximadamente 0,2) en pacientes con Mutaciones en SLC39A13 [Giunta et al., 2008]. Este método rápido y rentable también puede usarse para determinar el estado patogénico de un VUS (ver también «verificación del diagnóstico» en SEDk-PLOD1).
La ausencia de hallazgos genéticos confirmativos no excluye el diagnóstico de SEDsp, ya que los tipos específicos de mutaciones (por ejemplo, mutaciones intrónicas profundas) pueden pasar desapercibidos por las técnicas de diagnóstico molecular estándar, y aún otros, aún no descubiertos, pueden estar asociados con estos fenotipos. En caso de que no se identifiquen mutaciones de B4GALT7, B3GALT6 o SCL39A13, se deben considerar diagnósticos alternativos.
SED musculocontractural (SEDmc)
- Herencia
Autosómica recesiva
- Criterios mayores
-
Las contracturas múltiples congénitas, característicamente contracciones de aducción-flexión y/o talipes equinovarus (pie zambo)
-
Rasgos característicos craneofaciales, que son evidentes al nacer o en la primera infancia 38
-
Rasgos cutáneos característicos, incluyendo hiperextensibilidad de la piel 39, fragilidad de la piel con cicatrices atróficas, arrugas palmares aumentadas
- Criterios menores
-
Dislocaciones recurrentes/crónicas 40
-
Deformidades del pectum (plano, excavado)
-
Deformidades espinales (escoliosis, cifoscoliosis)
-
Dedos peculiares (estrechos, delgados, cilíndricos)
-
Deformidades progresivas de los pies (valgus, planus, cavum)
-
Hematomas subcutáneos grandes
-
Estreñimiento crónico
-
Divertículos colónicos
-
Pneumotórax/neumohemotórax
-
Nefrolitiasis/cistolitiasis
-
Hidronefrosis
-
Criptorquidia en los hombres
-
Estrabismo
-
Errores refractivos (miopía, astigmatismo)
-
Glaucoma / presión intraocular elevada
- Criterios mínimos sugerentes para los SEDmc
-En el nacimiento o en la primera infancia: criterios mayores (1)
– Contracturas congénitas múltiples Y (2) Rasgos craneofaciales característicos
– En la adolescencia y en la edad adulta: Criterios mayores (1)
– Contracturas congénitas múltiples Y (3) Rasgos cutáneos característicos
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
- Base Molecular
El SEDmc es causado por mutaciones bialélicas en CHST14, que codifica D4ST1, un gen de un solo exón que codifica la carbohidrato sulfotransferasa 14 o dermatan 4-O-sulfotransferasa 1, una enzima implicada en la biosíntesis del dermatan sulfato GAG. Cataliza la 4-O-sulfatación de N-acetilgalactosamina (GalNAc) en la secuencia «ácido L-idurónico (IdoA) -GalNAc» inmediatamente después de la epimerización del ácido D-glucurónico (GlcA) a IdoA por dermatosulfato epimerasa (DSE).
Se han identificado algunas mutaciones en el gen DSE, que codifica DSE, en pacientes con un fenotipo similar.
- Verificación del diagnóstico
Está indicada la detección molecular mediante resecuenciación dirigida de un panel genético que incluye CHST14 y DSE. Dicho panel genético también incluye otros genes asociados con fenotipos que se superponen clínicamente con mcEDS, tales como PLOD1, FKBP14, ZNF469, PRDM5, B4GALT7, B3GALT6 y SLC39A13. Alternativamente, el WES puede ser realizado. Cuando ninguna, o sólo una, mutación causante se identifica, este enfoque debe complementarse con una estrategia de detección de CNV para identificar grandes deleciones o duplicaciones en estos genes.
La ausencia de estos hallazgos genéticos confirmatorios no excluye el diagnóstico de SEDmc, ya que tipos específicos de mutaciones (por ejemplo, mutaciones intrónicas profundas) pueden pasar desapercibidos por las técnicas moleculares de diagnóstico estándar. En caso de que no se identifiquen mutaciones CHST14 o DSE, se deben considerar diagnósticos alternativos.
“El SEDmc es causado por mutaciones bialélicas en CHST14, que codifica D4ST1, un gen de un solo exón que codifica la carbohidrato sulfotransferasa 14 o dermatan 4-O-sulfotransferasa 1, una enzima implicada en la biosíntesis del dermatan sulfato GAG”.
SED miopático (SEDm)
- Herencia
Autosómica dominante o autosómica recesiva
- Criterios mayores
-
Hipotonía muscular congénita y/o atrofia muscular, que mejora con la edad41
-
Contracturas de las articulaciones proximales (rodilla, cadera y codo) 42
-
Hipermovilidad de las articulaciones distales
- Criterios menores
-
-Piel suave y pastosa
-
-Cicatrices atróficas
-
-Retraso del desarrollo motor
-
-Miopatía en la biopsia muscular
- Criterios clínicos mínimos sugerentes para SEDm
– Criterio mayor (1): hipotonía congénita del músculo que mejora con la edad
Más
– otro criterio mayor
– Y/o: tres criterios menores
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
- Base molecular
El SEDm es causada por mutaciones heterocigóticas o bialélicas en COL12A1, que codifica el colágeno tipo XII. El fenotipo clínico se superpone con las miopatías relacionadas con el colágeno tipo VI, es decir, la miopatía de Bethlem y la distrofia muscular congénita de Ullrich. Actualmente se desconoce si otros genes aún no descubiertos están asociados con este fenotipo.
- Verificación del diagnóstico
Está indicada la detección molecular mediante resecuenciación dirigida de un panel genético que incluye COL12A1. Dicho panel genético también incluye otros genes asociados con fenotipos que se superponen clínicamente con SEDm, tales como COL6A1, COL6A2, COL6A3. Alternativamente, el WES puede ser realizado. Cuando ninguna, o sólo una, mutación causante se identifica, este enfoque debe complementarse con una estrategia de detección de CNV para identificar grandes deleciones o duplicaciones en estos genes.
La ausencia de estos hallazgos confirmatorios no excluye el diagnóstico, ya que tipos específicos de mutaciones (por ejemplo, mutaciones intrónicas profundas) pueden pasar desapercibidos por las técnicas moleculares de diagnóstico estándar, y otros, aún no descubiertos, pueden estar asociados con este fenotipo. En caso de que no se identifiquen mutaciones de COL12A1, se deben considerar otros diagnósticos, especialmente la distrofia muscular congénita de Ullrich y la miopatía de Bethlem, relacionadas con el colágeno VI.
SED periodontal (SEDp)
- Herencia
Autosómica dominante
- Criterios mayores
-
Periodontitis persistente e intratable de inicio precoz (infancia o adolescencia)
-
Falta de encías adheridas
-
Placas pretibiales
-
Historia familiar de un familiar de primer grado que cumpla con los criterios clínicos
- Criterios menores
-
Propensión a los hematomas
-
Hipermovilidad articular, principalmente articulaciones distales
-
Hiperextensibilidad y fragilidad de la piel 40, cicatrices anormales (anchas o atróficas)
-
Aumento de la tasa de infecciones
-
Hernias
-
Rasgos faciales marfanoides
-
Acrogeria
-
Vasculatura prominente
- Criterios mínimos sugerentes pare SEDp
– Criterio mayor (1): periodontitis severa e intratable de inicio precoz (infancia o adolescencia)
– O Criterio mayor (2): falta de encía adherida
Más
– Al menos otros dos criterios mayores y un criterio menor
La prueba molecular confirmatoria es obligatoria para llegar a un diagnóstico final.
- Base molecular
SEDp es causada por mutaciones de ganancia de función heterocigóticas en C1R o C1S, codificando las subunidades C1r y C1s del primer componente de la vía clásica del complemento.
- Verificación del diagnóstico
La identificación de mutaciones conocidas o compatibles por análisis de secuencia de C1R y C1S. Las grandes deleciones o mutaciones nulas que eliminan completamente la función de proteína C1r o C1s no causan SEDp.
En la actualidad no se puede establecer si la ausencia de mutaciones C1R o C1S excluye el diagnóstico porque la experiencia con el diagnóstico molecular es limitado.
OBSERVACIONES FINALES
Esperamos que los nuevos criterios SED internacionales sirvan como una nueva, aunque provisional, norma para el diagnóstico de SED. Nuestra propuesta tiene como objetivo facilitar diagnósticos precisos y oportunos y mejorar la uniformidad diagnóstica para fines clínicos y de investigación, asesoramiento genético, abordaje, estudios de historia natural e identificación de posibles áreas de investigación. La futura revisión de esta Clasificación SED se planificará dentro del marco del Consorcio Internacional SED y de la Sociedad Ehlers-Danlos.
AGRADECIMIENTOS
Estamos muy agradecidos por la cooperación de muchos pacientes y sus familias en todo el mundo. Agradecemos al profesor Stephen Robertson y al apoyo de PVNH por su ayuda con PVNH-SED. También deseamos reconocer las contribuciones de muchos profesionales a este esfuerzo. Quisiéramos agradecer a nuestros generosos patrocinadores incluyendo, pero no limitados a, The Ehlers-Danlos National Foundation, the Ehlers-Danlos UK Support, y the Ehlers-Danlos Society. Fransiska Malfait es Investigadora Clínica Senior del Fondo de Investigación Científica de Flandes.
Notas finales
-
La extensibilidad de la piel se debe medir pinchando y elevando las capas cutánea y subcutánea de la piel en la superficie volar en la mitad del antebrazo no dominante como se describe en Remvig et al. [2009]. La piel es hiperextensible si se puede estirar sobre un corte estandarizado en tres de las siguientes áreas: 1,5 cm para la parte distal de los antebrazos y el dorso de las manos; 3 cm para el cuello, codo y rodillas.
-
Las cicatrices anormales pueden variar en gravedad. La mayoría de los pacientes tienen cicatrices atróficas extensas en varios sitios (Fig. 1). Éstos pueden ser a veces hemosideróticos. Una minoría de pacientes son más ligeramente afectados.
-
HAG se evalúa según la puntuación de Beighton; Una puntuación de Beighton ? 5 se considera positiva para la presencia de HAG (Fig. 2). Dado que la laxitud disminuye con la edad, los pacientes con un puntaje de Beighton <5/9 pueden ser considerados positivos basándose en sus observaciones históricas (ver «cuestionario de cinco puntos (5PQ)» (Tabla III).
-
El moretón fácil puede ocurrir en cualquier parte del cuerpo, incluyendo sitios inusuales. El área pretibial a menudo permanece manchada con hemosiderina de moretones anteriores.
-
La alteración subjetiva de la textura de la piel es apreciable al tocar la piel.
-
Los pseudotumores moluscoides son lesiones carnudas asociadas con cicatrices, que se encuentran sobre puntos de presión (por ejemplo, codo, dedos).
-
Los esferoides subcutáneos (Fig. 1E) son pequeños cuerpos esféricos duros, frecuentemente móviles y palpables en los antebrazos y las espinillas. Los esferoides pueden calcificarse y detectarse radiológicamente.
-
Los pliegues epicantales se observan a menudo en la infancia, pero también pueden observarse en adultos.
-
Para las definiciones de HAG y hiperextensibilidad de la piel, vea los criterios para «SED clásico».
-
Los problemas cardíacos-valvulares se informaron en todos los individuos adultos afectados, pero estaban ausentes en los dos niños reportados (ambos <10 años de edad).
-
Para la definición de la hiperextensibilidad de la piel, véase criterios para «SED clásico».
-
Si existen características marfanoides, considere otras afecciones como: síndrome de Marfan, síndrome de Loeys-Dietz, arcnodactilia contractural congénita, síndrome de Shprintzen-Goldberg, síndrome de Stickler, homocistinuria, neoplasia endocrina múltiple tipo 2B y trastornos aneurismáticos aórticos torácicos familiares [Pyeritz y Loeys, 2012]. Las pruebas moleculares para muchas de estas condiciones están clínicamente disponibles.
-
Mientras que la suavidad y la textura de la piel permanecen subjetivas, a menudo es muy notable en algunos individuos y útil cuando está presente pero no cuantificable; Por lo tanto recomendamos un umbral alto para la positividad.
-
Extensibilidad de la piel medida por pellizcar y levantar las capas cutánea y subcutánea de la piel en la superficie volar en la mitad del antebrazo no dominante como se describe en Remvig et al. [2009]. La extensibilidad cutánea> 1,5 cm se considera el extremo superior de la normalidad. Es probable que la hiperextensibilidad de la piel en los SEDh se sobreponga significativamente con la de la piel «normal». Por lo tanto, la extensibilidad de más de 1,5 cm es «positiva». Si la extensibilidad> 2,0 cm está presente especialmente en combinación con otras características cutáneas, tales como cicatrices papiráceas, pseudotumores moluscoides y / o esferoides subcutáneos, considere otros tipos de SED como posibles diagnósticos alternativos. Principalmente SEDc y SEDcl).
-
Las pápulas piezógenas son hernias de grasa subcutánea a menudo demostrables en el talón al estar de pie (Fig. 3). Sin embargo, en un estudio de 29 pacientes holandeses con SED en edad y sexo, se consideró poco frecuente en niños, pero puede encontrarse en adultos con antecedentes de bipedestación prolongada (ocupacional), corredores de maratón o levantadores de pesas [Poppe y Hamm, Se encontraron pápulas piezogénicas en el 34,5%, pero ninguna en el grupo control [Kahana et al., 1987].
-
Las cicatrices atróficas se definen como cicatrices de laceraciones traumáticas lineales o de cirugía única que son inusualmente superficiales (es decir, delgadas y hundidas) y/o más anchas que la herida original debido a alteración de la reparación y posterior hipotrofia dérmica. Las cicatrices atróficas como resultado de incisiones múltiples, infecciones de la herida o condiciones inflamatorias (tales como infecciones virales, acné quístico, etc.) no deben considerarse. Las incisiones elípticas (por ejemplo, para la eliminación de nevus) pueden ser difíciles de evaluar sin conocer el tamaño de la herida original. La verdadera fragilidad de la piel, como la propensión a tener una herida abierta debido a un trauma trivial, no es una característica típica de los SEDh. La cicatrización atrófica en los SEDh es ligeramente a moderadamente diferente de la que usualmente se considera típica de SEDc (Fig. 1).
-
Incluye historia de aglomeración dental o intervención de ortodoncia para corregir tales problemas. Ambas condiciones deben ser positivas para cumplir este criterio.
-
Algunos estudios no muestran aumento en la frecuencia de MVP clínicamente significativo [Dolan et al., 1997; McDonnell et al., 2006; Atzinger et al., 2011] y otros muestran una frecuencia MVP de 28-67% entre los pacientes con SEDh [Camerota et al., 2014; Kozanoglu et al., 2016]. Esta característica se incluye en los criterios de diagnóstico porque puede ser un marcador de laxitud de tejido conectivo, pero suele ser significativa en pacientes con SEDh.
-
«Dislocación» se define como el desplazamiento de un hueso fuera de la cavidad articular (o fuera de su posición normal en el caso de huesos sesamoideos como la rótula), lo suficientemente severo para limitar el movimiento de la articulación y requerir una reducción manual.
-
Se refiere a los sitios independientemente de la lateralidad. Por ejemplo, la inestabilidad patelar derecha e izquierda contaría como dos. La inestabilidad debe ser evaluada y determinada por un profesional calificado usando las pautas recomendadas.
-
Todos los pacientes con SEDa presentaron dislocación bilateral congénita de cadera. Se sabe que un paciente de SEDa no reportado y molecularmente probado tuvo una luxación unilateral congénita de cadera [Byers et al., Comunicación personal].
-
Para la definición de HAG, véase criterios para «SED clásico».
-
Los rasgos craneofaciales incluyen: ojos prominentes y protuberantes con párpados hinchados y edematosos y piel periorbitaria excesiva, pliegues epicánticos, fisuras palpebrales descendentes, escleras azules, grandes fontanelas y/o anchas suturas craneales, cierre tardío de las fontanelas y barbilla hipoplásica.
-
Las complicaciones perinatales reportadas debido a la fragilidad del tejido conectivo incluyen: fracturas congénitas del cráneo, hemorragia intracerebral, cordón umbilical friable, desgarros congénitos de la piel, neumotórax neonatal.
-
Para la definición de HAG, véase criterios para «SED clásica».
-
La mayoría de los pacientes identificados hasta la fecha muestran un fenotipo grave, reconocible desde el nacimiento o primeros meses de vida. Recientemente se han descrito formas más leves de la enfermedad.
-
La hipotonía muscular puede ser muy pronunciada y conducir al retraso de desarrollo motor grueso. Esta condición debe considerarse en el diagnóstico diferencial inicial de un niño flexible. Sin embargo, el tratamiento neuromuscular es normal.
-
La cifoscoliosis suele estar presente al nacer o se desarrolla en la infancia. En pacientes con mutaciones bialélicas PLOD1, puede estar ausente durante la edad adulta.
-
Para las definiciones de HAG y hiperextensibilidad de la piel, vea los criterios para «SED clásico».
-
La fragilidad escleral y ocular se eliminó de los Criterios mayores clínicos de SEDk-PLOD1, ya que la ruptura del globo ocular después de un traumatismo mínimo sólo se ha reportado en cinco individuos, incluyendo un paciente con ambos ojos afectados.
-
Las características dismórficas faciales incluyen: orejas de bajo ajuste, pliegues epicánticos, fisuras palpebrales inclinadas hacia abajo, sinofris y paladar alto.
-
Para las definiciones de HAG y hiperextensibilidad de la piel, vea los criterios para «SED clásico».
-
Los rasgos craneofaciales característicos de las mutaciones bialélicas B4GALT7 incluyen: cara triangular, ojos ampliamente espaciados, proptosis, boca estrecha, orejas de baja altura, pelo escaso del cuero cabelludo, dentición anormal, cara plana, frente ancha, escleras azules y paladar hendido .
-
Los hallazgos radiográficos notificados asociados con las mutaciones bialélicas B4GALT7 incluyen: incluyen sinostosis radioulnar, ensanchamiento metafisario, osteopenia, subluxación o luxación radial de la cabeza y clavículas cortas con extremos medianos anchos.
-
Los rasgos craneofaciales característicos de las mutaciones bialélicas B3GALT6 incluyen: hipoplasia de la parte media de la cara, protuberancia frontal, proptosis o ojos prominentes, escleróticas azules, fisuras palpebrales descendentes, puente nasal deprimido, orificios bajos, micrognatia, dentición anormal, paladar hendido
-
Las características radiográficas asociadas con las mutaciones bialélicas B3GALT6 incluyen: platispondilia, pico anterior del cuerpo vertebral, ilium corto, trocanter prominente menor, displasia acetabular, ensanchamiento metafisario, displasia metafisaria de la cabeza femoral, desalinieamiento del codo, dislocación de la cabeza radial, tubulacion excesiva, arqueamiento de los huesos largos , Osteoporosis generalizada, fracturas cicatrizadas. Se ha reportado craneosinostosis y disostosis radioulnar en un paciente.
-
Los hallazgos radiológicos notificados asociados con las mutaciones bialélicas SLC39A13 incluyen: ligera a moderada platispondilia, osteopenia leve a moderada de la columna vertebral, ileo pequeño, epífisis proximal femoral plana, cuello femoral corto y ancho.
-
Los rasgos craneofaciales característicos: fontanela grande, hipertelorismo, fisuras palpebrales cortas y descendentes, escleróticas azules, nariz corta con columela hipoplásica, orejas de baja fijación y rotación, paladar alto, filtrado largo, bermellón del labio superior delgado, boca pequeña, micro-retrognatia.
-
Para la definición de la hiperextensibilidad de la piel, véase criterios para «SED clásico».
-
Las características fenotípicas en los tres pacientes reportados con SED causada por deficiencia de DSE parecen ser más leves que en pacientes con SED causada por la deficiencia de D4ST1, pero la identificación de pacientes adicionales con deficiencia de DSE es necesaria para confirmar esta correlación.
-
Hasta el momento, se han reportado cinco familias: cuatro con una condición autosómica dominante y otra con una enfermedad autosómica recesiva. Los hermanos afectados de la familia con la condición autosómica recesiva tienen una forma más severa de la enfermedad que los pacientes con herencia autosómica dominante [Zou et al., 2014].
-
Biopsia muscular y estudios de cultivo de fibroblastos de la piel: el diagnóstico puede sospecharse en pacientes que se someten a una biopsia muscular y/o en los que se establece una línea de fibroblastos. En la forma autosómica recesiva en la que no se produce colágeno XII, la inmunotinción ha mostrado ausencia de tinción con colágeno XII. En las mutaciones erróneas que conducen a formas autosómicas dominantes, el colágeno XII puede ser anormalmente secretado. El patrón miopático en la biopsia muscular puede ser sugestivo, pero no es diagnóstico. Recientemente, la resonancia magnética muscular se ha desarrollado como una técnica alternativa no invasiva para estudiar la afectación muscular, sin embargo, no es lo suficientemente específica como para confirmar el diagnóstico.
REFERENCIAS/Bibliografía
Atzinger CL, Meyer RA, Khoury PR, Gao Z, Tinkle BT. 2011. Cross-sectional and longitudinal assessment of aortic root dilation and valvular anomalies in hypermobile and classic Ehlers-Danlos syndrome. J Pediatr 158:826–830 e821.
Baumann M, Giunta C, Krabichler B, Ruschendorf F, Zoppi N, Colombi M, Bittner RE, Quijano-Roy S, Muntoni F, Cirak S, Schreiber G, Zou Y, Hu Y, Romero NB, Carlier RY, Amberger A, Deutschmann A, Straub V, Rohrbach M, Steinmann B, Rostasy K, Karall D, Bonnemann CG, Zschocke J, Fauth C. 2012. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. Am J Hum Genet 90:201–216.
Beighton P, de Paepe A, Danks D, Finidori G, Gedde-Dahl T, Goodman R, Hall JG, Hollister DW, Horton W, McKusick VA, et al. 1988. International nosology of heritable disorders of connective tissue, Berlin, 1986. Am J Med Genet 29:581–594.
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. 1998. Ehlers-Danlos syndromes: Revised nosology, villefranche, 1997. ehlers-Danlos national foundation (USA) and ehlers-Danlos support group (UK). Am J Med Genet 77:31–37.
Camerota F, Castori M, Celletti C, Colotto M, Amato S, Colella A, Curione M, Danese C. 2014. Heart rate, conduction and ultrasound abnormalities in adults with joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Clin Rheumatol 33:981–987.
Dolan AL, Mishra MB, Chambers JB, Grahame R. 1997. Clinical and echocardiographic survey of the Ehlers-Danlos syndrome. Br J Rheumatol 36:459–462.
Giunta C, Elçioglu NH, Albrecht B, Eich G, Chambaz C, Janecke AR, Yeowell H, Weis M, Eyre DR, Kraenzlin M, Steinmann B. 2008. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome—An autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am J Hum Genet 82:1290–1305.
Hakim AJ, Grahame R. 2003. A simple questionnaire to detect hypermobility: An adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract 57:163–166.
Hautala T, Heikkinen J, Kivirikko KI, Myllyla R. 1993. A large duplication in the gene for lysyl hydroxylase accounts for the type VI variant of Ehlers-Danlos syndrome in two siblings. Genomics 15:399–404.
Kahana M, Feinstein A, Tabachnic E, Schewach-Millet M, Engelberg S. 1987. Painful piezogenic pedal papules in patients with Ehlers-Danlos syndrome. J Am Acad Dermatol 17:205–209.
Kozanoglu E, Coskun Benlidayi I, Akilli R, Tasal A. 2016. Is there any link between joint hypermobility and mitral valve prolapse in patients with fibromyalgia syndrome? Clin Rheumatol 35:1041–1044.
Malfait F, Symoens S, Coucke P, Nunes L, De Almeida S, De Paepe A. 2006. Total absence of the alpha2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity to cardiac valvular problems. J Med Genet 43:e36.
Malfait F, Symoens S, De Backer J, Hermanns-Le T, Sakalihasan N, Lapiere CM, Coucke P, De Paepe A. 2007a. Three arginine to cysteine substitutions 4 in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Human Mutation 28:387–395.
Malfait F, Symoens S, De Backer J, Hermanns-Le T, Sakalihasan N, Lapiere CM, Coucke P, De Paepe A. 2007b. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat 28:387–395.
McDonnell NB, Gorman BL, Mandel KW, Schurman SH, Assanah-Carroll A, Mayer SA, Najjar SS, Francomano CA. 2006. Echocardiographic findings in classical and hypermobile Ehlers-Danlos syndromes. Am J Med Genet A 140A:129–136.
McKay MJ, Baldwin JN, Ferreira P, Simic M, Vanicek N, Hiller CE, Nightingale EJ, Moloney NA, Quinlan KG, Pourkazemi F, Sman AD, Nicholson LL, Mousavi SJ, Rose K, Raymond J, Mackey MG, Chard A, Hubscher M, Wegener C, Fong Yan A, Refshauge KM, Burns J, Norms Project C. 2016. 1000 Norms Project: Protocol of a cross-sectional study cataloging human variation. Physiotherapy 102:50–56.
Mulvey MR, Macfarlane GJ, Beasley M, Symmons DP, Lovell K, Keeley P, Woby S, McBeth J. 2013. Modest association of joint hypermobility with disabling and limiting musculoskeletal pain: Results from a large-scale general population-based survey. Arthritis Care Res (Hoboken) 65:1325–1333.
Poppe H, Hamm H. 2013. Piezogenic papules in Ehlers-Danlos syndrome. J Pediatr 163:1788.
Pyeritz RE, Loeys B. 2012. The 8th international research symposium on the Marfan syndrome and related conditions. Am J Med Genet A 158A:42–49.
Remvig L, Duhn PH, Ullman S, Kobayasi T, Hansen B, Juul-Kristensen B, Jurvelin JS, Arokoski J. 2009. Skin extensibility and consistency in patients with Ehlers-Danlos syndrome and benign joint hypermobility syndrome. Scand J Rheumatol 38:227–230.
Remvig L, Jensen DV, Ward RC. 2007. Are diagnostic criteria for general joint hypermobility and benign joint hypermobility syndrome based on reproducible and valid tests? A review of the literature. J Rheumatol 34:798–803.
Ritelli M, Dordoni C, Venturini M, Chiarelli N, Quinzani S, Traversa M, Zoppi N, Vascellaro A, Wischmeijer A, Manfredini E, Garavelli L, Calzavara-Pinton P, Colombi M. 2013. Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: Identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet J Rare Dis 8:58.
Rohrbach MSH, Porter LF, Burkitt-Wright EM, Bürer C, Janecke A, Bakshi M, Sillence D, Al-Hussain H, Baumgartner M, Steinmann B, Black GC, Manson FD, Giunta C. 2013. ZNF469 frequently mutated in the brittle cornea syndrome (BCS) is a single exon gene possibly regulating the expression of several extracellular matrix components. Mol Genet Metab 109:289–295.
Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, van Haren B, Miller WL, Bristow J. 2001. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med 345:1167–1175.
Schwarze U, Hata R, McKusick VA, Shinkai H, Hoyme HE, Pyeritz RE, Byers PH. 2004. Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway. Am J Hum Genet 74:917–930.
Smits-Engelsman B, Klerks M, Kirby A. 2011. Beighton score: A valid measure for generalized hypermobility in children. J Pediatr 158:119–123 123 e111-114.
Soucie JM, Wang C, Forsyth A, Funk S, Denny M, Roach KE, Boone D, Hemophilia Treatment Center N. 2011. Range of motion measurements: Reference values and a database for comparison studies. Haemophilia 17:500–507.
Symoens S, Syx D, Malfait F, Callewaert B, De Backer J, Vanakker O, Coucke P, De Paepe A. 2012. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum Mutat 33:1485–1493.
Talhaoui I, Bui C, Oriol R, Mulliert G, Gulberti S, Netter P, Coughtrie MW, Ouzzine M, Fournel-Gigleux S. 2010. Identification of key functional residues in the active site of human {beta}1, 4-galactosyltransferase 7: A major enzyme in the glycosaminoglycan synthesis pathway. J Biol Chem 285:37342–37358.
Weerakkody RA, Vandrovcova J, Kanonidou C, Mueller M, Gampawar P, Ibrahim Y, Norsworthy P, Biggs J, Abdullah A, Ross D, Black HA, Ferguson D, Cheshire NJ, Kazkaz H, Grahame R, Ghali N, Vandersteen A, Pope FM, Aitman TJ. 2016. Targeted next-generation sequencing makes new molecular diagnoses and expands genotype-phenotype relationship in Ehlers-Danlos syndrome. Genet Med 18:1119–1127.
Zoppi N, Chiarelli N, Cinquina V, Ritelli M, Colombi M. 2015. GLUT10 deficiency leads to oxidative stress and non-canonical alphavbeta3 integrin-mediated TGFbeta signalling associated with extracellular matrix disarray in arterial tortuosity syndrome skin fibroblasts. Hum Mol Genet 24:6769–6787.
Zou Y, Zwolanek D, Izu Y, Gandhy S, Schreiber G, Brockmann K, Devoto M, Tian Z, Hu Y, Veit G, Meier M, Stetefeld J, Hicks D, Straub V, Voermans NC, Birk DE, Barton ER, Koch M, Bonnemann CG 2014. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum Mol Genet 23:2339–2352
Fuente:
American Journal of Medical Genetics Part C (Seminars in Medical Genetics)
Traducción (Julio 2017):
Aixa Toro Zarca
Autores:
Fransiska Malfait Clair Francomano Peter Byers John Belmont Britta Berglund James Black Lara Bloom Jessica M. Bowen Angela F. Brady Nigel P. Burrows Marco Castori Helen Cohen Marina Colombi Serwet Demirdas Julie De Backer Anne De Paepe Sylvie Fournel?Gigleux Michael Frank Neeti Ghali Cecilia Giunta Rodney Grahame Alan Hakim Xavier Jeunemaitre Diana Johnson Birgit Juul?Kristensen Ines Kapferer?Seebacher Hanadi Kazkaz Tomoki Kosho Mark E. Lavallee Howard Levy Roberto Mendoza?Londono Melanie Pepin F. Michael Pope Eyal Reinstein Leema Robert Marianne Rohrbach Lynn Sanders Glenda J. Sobey Tim Van Damme Anthony Vandersteen Caroline van Mourik Nicol Voermans Nigel Wheeldon Johannes Zschocke Brad Tinkle